ニーマン・ピック病
ニーマン・ピック病(NPD)は、脾臓・肝臓・脳の細胞に脂質と呼ばれる脂肪物質が蓄積する家系(遺伝性)を経て受け継がれる疾患群です。
この病気には 3 つの一般的な形態があります。
- タイプA
- タイプB
- タイプC
それぞれのタイプは、異なる器官を含みます。神経系や呼吸に関係する場合とそうでない場合があります。それぞれが異なる症状を引き起こす可能性があり、生涯を通じて異なる時期に発生する可能性があります。
NPD タイプ A および B は、体内の細胞が酸性スフィンゴミエリナーゼ (ASM) と呼ばれる酵素を持たない場合に発生します。この物質は、体内のすべての細胞に見られるスフィンゴミエリンと呼ばれる脂肪物質の分解 (代謝) を助けます。
ASM が不足していたり、正常に機能していない場合、細胞内にスフィンゴミエリンが蓄積します。これにより細胞が死滅し、臓器が適切に機能しにくくなります。
タイプ A は、すべての人種と民族に発生します。アシュケナージ (東欧) のユダヤ人に多く見られます。
C 型は、体がコレステロールやその他の脂肪 (脂質) を適切に分解できない場合に発生します。これにより、肝臓と脾臓でコレステロールが過剰になり、脳内の他の脂質が過剰になります。タイプCは、スペイン系プエルトリコ人の間で最も一般的です。
タイプ C1 はタイプ C の変形です。これは、コレステロールが脳細胞間を移動する方法を妨げる欠陥を伴います。このタイプは、ノバスコシア州ヤーマス郡のフランス系カナダ人にのみ見られます。
症状はさまざまです。他の健康状態が同様の症状を引き起こす可能性があります。病気の初期段階では、いくつかの症状しか引き起こさない場合があります。人によってすべての症状が現れることはありません。
A型は通常、生後数か月以内に始まります。症状には次のようなものがあります。
- 3~6ヶ月で腹部(お腹周り)の腫れ
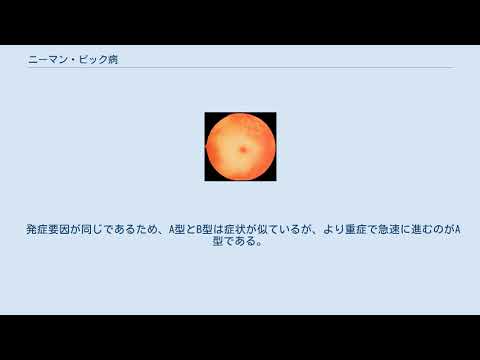
- 目の後ろ(網膜上)にあるチェリーレッドの斑点
- 摂食困難
- 初期の運動能力の喪失(時間の経過とともに悪化する)
B型の症状は通常軽度です。小児期後期または 10 代に発生します。乳幼児では腹部膨満が起こることがあります。運動能力の喪失など、脳や神経系の関与はほとんどありません。呼吸器感染症を繰り返す子供もいます。
C型とC1型は通常、学齢期の子供に影響を及ぼします。ただし、幼児期から成人期までのいつでも発生する可能性があります。症状には次のようなものがあります。
- 不安定な歩行、ぎこちない、歩行の問題につながる可能性がある手足の動きの困難
- 拡大した脾臓
- 肝腫大
- 出産時(または出産直後)の黄
- 学習困難と知的低下
- 発作
- 不明瞭で不規則な発話
- 転倒につながる可能性のある筋緊張の突然の喪失
- 震える
- 目を上下に動かすのが難しい
A型とB型を診断するために、血液検査や骨髄検査を行うことができます。この検査では、誰が病気にかかっているかはわかりますが、あなたが保因者かどうかはわかりません。 A型およびB型の保因者を診断するためにDNA検査を行うことができます。
皮膚生検は通常、C 型と D 型を診断するために行われます。医療提供者は、皮膚細胞がどのように成長、移動、コレステロールを蓄積するかを観察します。このタイプの病気の原因となる 2 つの遺伝子を探すために、DNA 検査が行われることもあります。
その他のテストには次のようなものがあります。
- 骨髄穿刺
- 肝生検(通常は不要)
- 細隙灯視力検査
- ASMのレベルをチェックするためのテスト
現在、A型に有効な治療法はありません。
B 型に対しては、骨髄移植が試みられる場合があります。研究者たちは、酵素補充療法や遺伝子治療など、可能な治療法を研究し続けています。
C型の神経症状には、ミグルスタットという新薬が登場しました。
高コレステロールは、健康的で低コレステロールの食事や薬で管理できます。ただし、研究は、これらの方法が病気の悪化を止めたり、細胞がコレステロールを分解する方法を変えたりすることを示していません。筋緊張の突然の喪失や発作など、多くの症状を制御または緩和するための薬が利用できます。
これらの組織は、ニーマン・ピック病に関するサポートと詳細情報を提供できます。
- 国立神経疾患・脳卒中研究所 -- www.ninds.nih.gov/Disorders/All-Disorders/Niemann-Pick-Disease-Information-Page
- 国立ニーマン・ピック病財団 -- nnpdf.org
- 全国稀な疾患のための機構 --希無病.org/rare-diseases/niemann-pick-disease-type-c
NPD タイプ A は、重篤な疾患です。通常、2 歳または 3 歳までに死亡します。
B型の人は、小児期後期または成人期まで生きることがあります。
1 歳になる前に C 型の徴候を示す子供は、学齢期まで生きることができません。入学後に症状が現れる人は、10代半ばから後半まで生きる可能性があります。 20代まで生きる人もいます。
ニーマン・ピック病の家族歴があり、子供を持つ予定がある場合は、医療提供者に予約してください。遺伝カウンセリングとスクリーニングが推奨されます。
お子様に次のようなこの病気の症状がある場合は、医療提供者に連絡してください。
- 発達上の問題
- 摂食障害
- 体重増加が不十分
すべてのタイプのニーマン・ピック病は常染色体劣性です。これは、両親が両方とも保因者であることを意味します。それぞれの親は、病気自体の兆候はなく、異常な遺伝子の 1 つのコピーを持っています。
両親が両方とも保因者の場合、子供が病気になる確率は 25%、子供が保因者になる確率は 50% です。
保因者検出検査は、遺伝的欠陥が特定された場合にのみ可能です。タイプ A および B に含まれる欠陥は十分に研究されています。これらの形態のニーマン・ピック病の DNA 検査が利用できます。
多くの C 型患者の DNA には遺伝的欠陥が確認されています。異常な遺伝子を持っている人の診断ができる可能性があります。
いくつかのセンターでは、まだ子宮内にある赤ちゃんを診断するための検査を提供しています。
NPD;スフィンゴミエリナーゼ欠損症;脂質貯蔵障害 - ニーマン・ピック病;リソソーム蓄積症 - ニーマン・ピック病
 ニーマン・ピック病の泡沫細胞
ニーマン・ピック病の泡沫細胞
Kliegman RM、St. Geme JW、Blum NJ、Shah SS、Tasker RC、Wilson KM..脂質代謝の欠陥。で:Kliegman RM、St. Geme JW、Blum NJ、Shah SS、Tasker RC、Wilson KM、eds。 ネルソン小児科の教科書。 第21版ペンシルバニア州フィラデルフィア: エルゼビア。 2020:104章
Turnpenny PD、Ellard S、Cleaver R. 先天性代謝異常。で: Turnpenny PD、Ellard S、Cleaver R、eds。 Emery の医療遺伝学およびゲノミクスの要素。 第 16 版ペンシルバニア州フィラデルフィア: エルゼビア。 2022:18章
興味深い記事

オプラの2019年のお気に入りのリストからのこれらの3つのグッズであなたの心と体を甘やかしてください